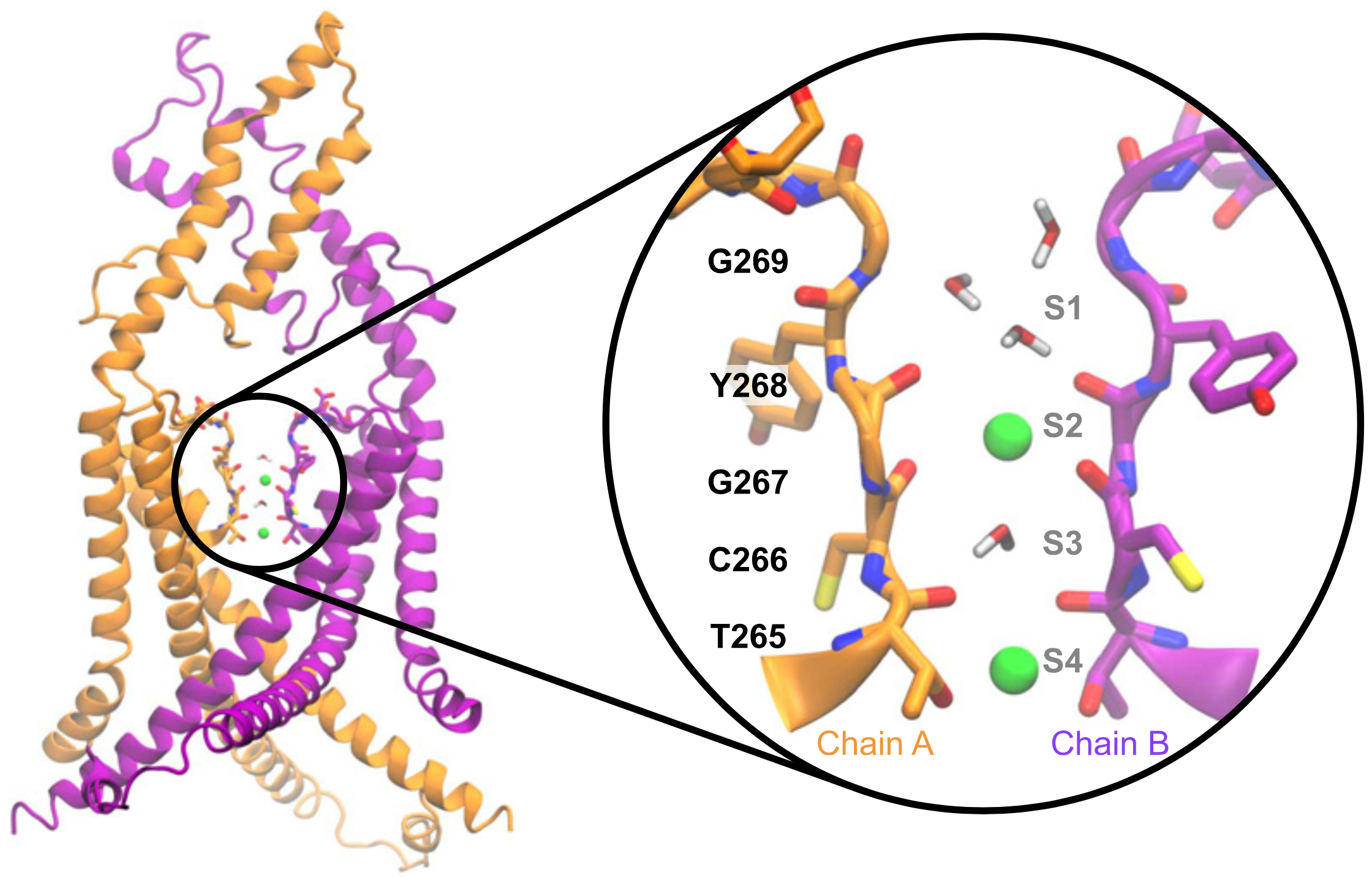
New publication in PNAS - Constitutive sodium permeability in a C. elegans two-pore domain potassium channel.
2024-10-15
Congratulations to all involved in this multi-disciplinary exploration of
the molecular determinants of K2P channel selectivity.
Potassium channels play a central role in modulating cellular excitability, particularly of neuronal cells. Their unique structure determines their ability to let ions pass selectively through cell membranes. The impact of pathological or evolutionary variations in this selectivity filter remains difficult to predict. Here, we reveal that UNC-58, a member of the two-pore domain potassium (K2P) channel family of C. elegans, exhibits an unusual sodium permeability due to a unique cysteine residue in its selectivity filter. Our findings underscore the importance of functional studies to determine how sequence variation in potassium channel selectivity filters can shape the electrical profiles of excitable cells.
New publication in Nature Communications - Wnt-Ror-Dvl signalling and the dystrophin complex organize planar-polarized membrane compartments in C. elegans muscles
2024-06-10
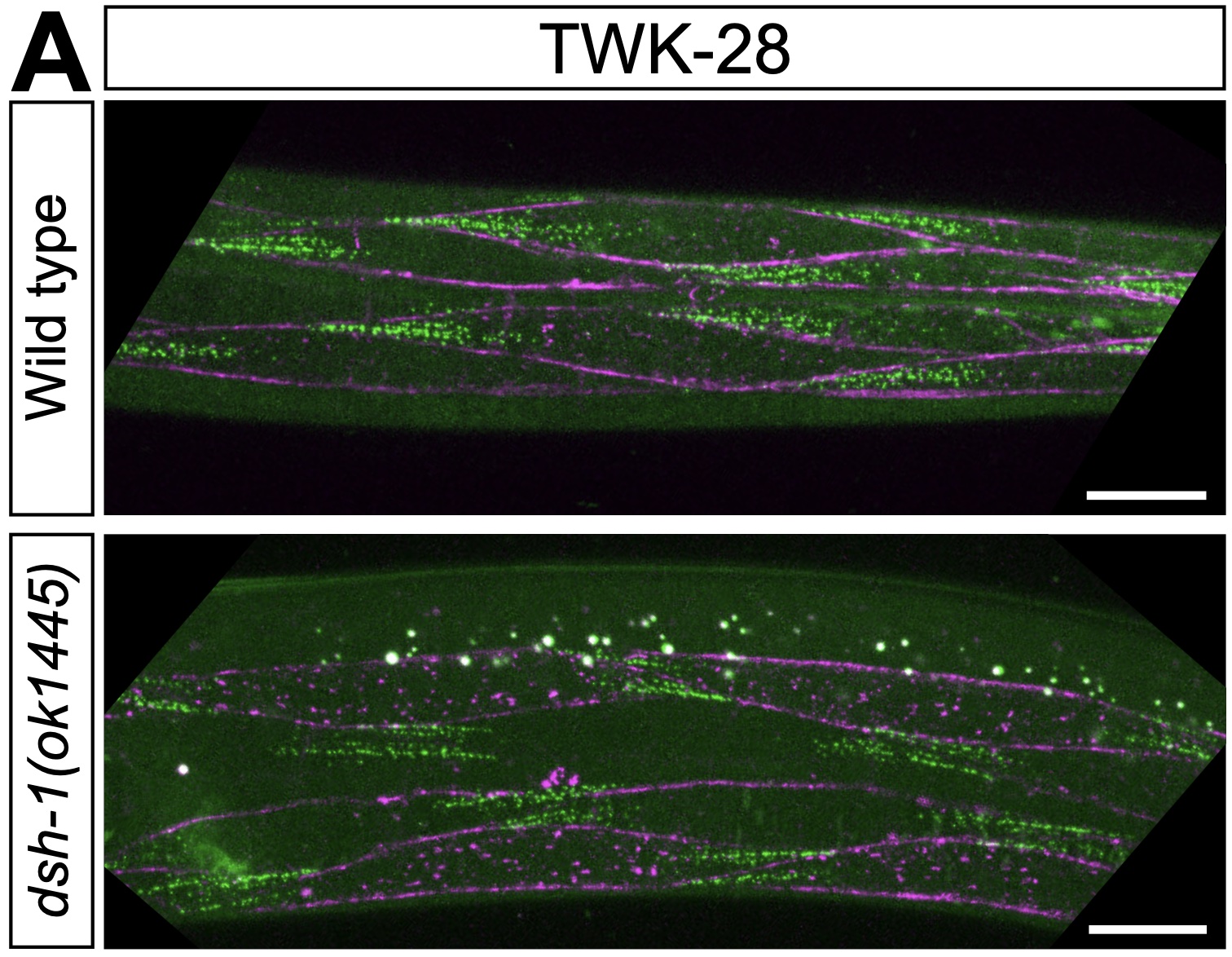
Our study revealing the remarkably complex organisation of the worm's sarcolemma is now out at
Nature Communications! Almost six years in the making, congratulations to Alice, Nora, Marie, Amandine, Noémie, Elise and Olga for this
magnum opus revealing this entirely unsuspected new case of
planar cell polarity in
C. elegans muscles.
SUMMARY
Cell polarity mechanisms allow the formation of specialized membrane domains with unique protein compositions, signalling properties, and functional characteristics. By analyzing the localization of potassium channels and proteins belonging to the dystrophin-associated protein complex, we reveal the existence of distinct planar-polarized membrane compartments at the surface of C. elegans muscle cells. We find that muscle polarity is controlled by a non-canonical Wnt signalling cascade involving the ligand EGL-20/Wnt, the receptor CAM-1/Ror, and the intracellular effector DSH-1/Dishevelled. Interestingly, classical planar cell polarity proteins are not required for this process. Using time-resolved protein degradation, we demonstrate that –while it is essentially in place by the end of embryogenesis– muscle polarity is a dynamic state, requiring continued presence of DSH-1 throughout post-embryonic life. Our results reveal the unsuspected complexity of the C. elegans muscle membrane and establish a genetically tractable model system to study cellular polarity and membrane compartmentalization in vivo.
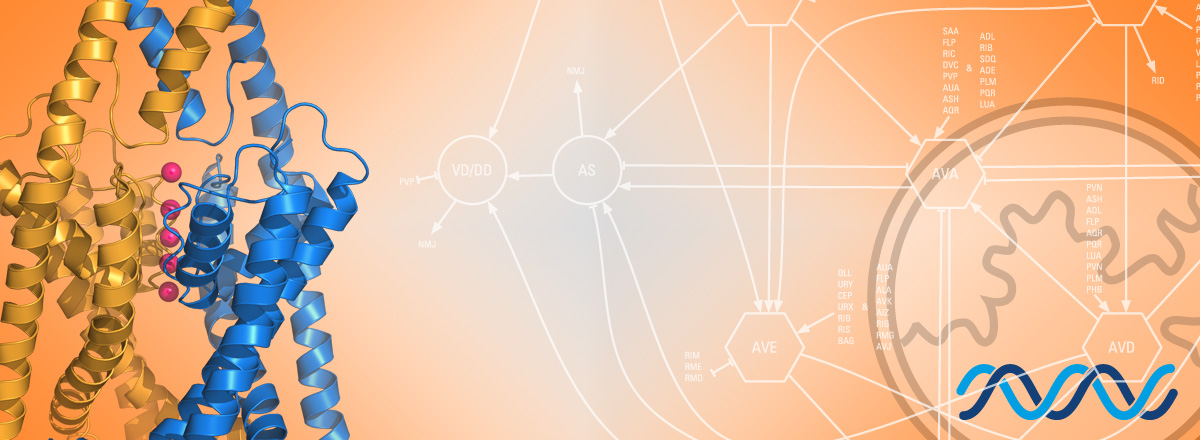
New publication in Science Advances - A tonically active master neuron modulates mutually exclusive motor states at two timescales
2024-04-10
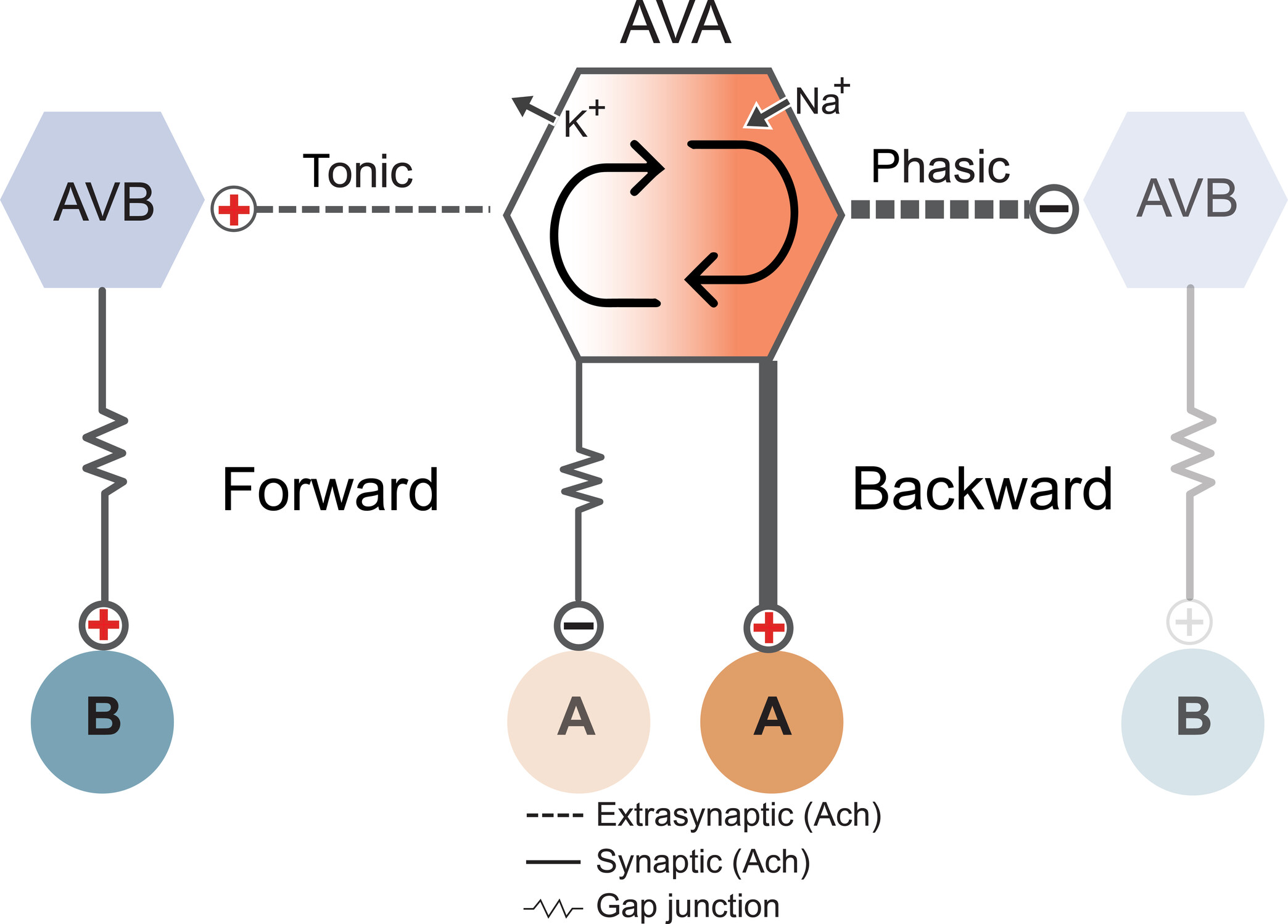
Congratulations to Jun, Sonia El Mouridi, and the teams of Mei Zhen and Shangbang Gao for our first collaborative study about
the exquisite regulation of C. elegans locomotion.
In this study, we report a new conceptual framework for a universal problem in motor control: how do animals generate smooth transitions when switching between opposing motor states like forward and backward locomotion? We analyzed circuits for forward and backward locomotion in C. elegans with single cell endogenous membrane potential perturbations, electrophysiology, calcium imaging, and mathematical modeling. In contrast to the prevailing view, our results establish a new model for smooth and continuous behavioral transitions between forward and backward locomotion.
New publication in eLife - Natural variation in the Caenorhabditis elegans egg-laying circuit modulates an intergenerational fitness trade-off
2024-04-02
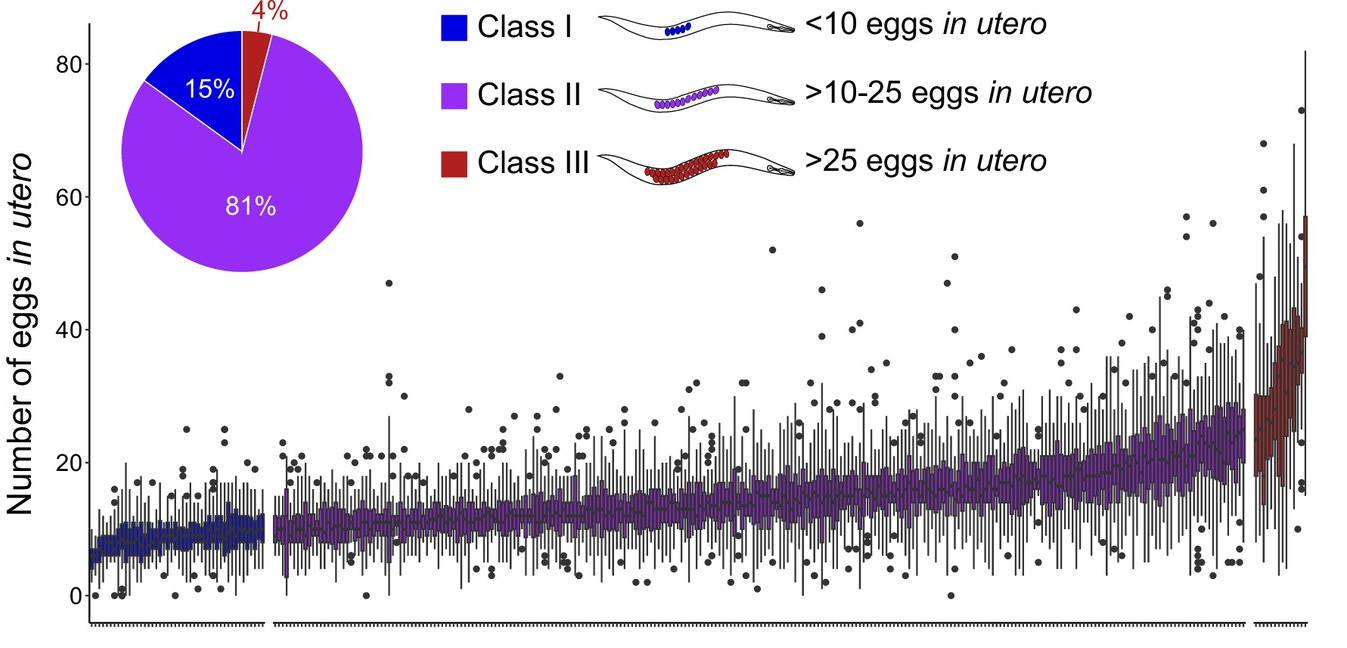
Congratulations to Laure Mignerot and the team of Christian Braendle for our second collaborative study about
the natural variation of the C. elegans egg-laying behaviour.
eLife assessment - This important work provides a thorough and detailed analysis of natural variation in C. elegans egg-laying behavior. The authors present convincing evidence to support their hypothesis that variations in egg-laying behavior are influenced by trade-offs between maternal and offspring fitness. This study establishes a framework for elucidating the molecular mechanisms underlying this paradigm of behavioral evolution.
NERVSPAN - Molecular plasticity of neurons during C. elegans lifespan
2024-05 - Marie Skłodowska-Curie Actions - European Training Network

Within the framework of the NERVSPAN European Doctoral Training Network, we are offering a fully-funded PhD fellowship to study the robustness of neuronal excitability and function across gender and lifespan.
The intrinsic electrical properties of neurons are defined by the repertoire of ion channels they express. Determining the precise genetic programs that establish the electrical identity of neurons remains a major question in cellular neuroscience. Single-cell transcriptomic studies in the C. elegans nervous system have revealed that potassium channels are often expressed in complex combinations of over a dozen channel genes in a single neuron. These different ion channel combinations will determine the biophysical and electrical properties defining neuron-specific functions throughout its life.
From a conceptual point of view, this project will address for the first time how ion channel degeneracy, variability and covariation are genetically regulated to ensure the resilience and phenotypic stability/plasticity of excitable cells in C. elegans.
NERVSPAN will train 12 doctoral Students at 7 academic institutions and 1 company. The nature of the programme will provide the enrolled students with strong interdisciplinary training (including, but not limited to: molecular biology, bioinformatics, high resolution microscopy, proteomics, and transcriptome profiling).
Find more information here.
New publication in Gene - Functional and clinical characterization of a novel homozygous KCNH2 missense variant in the pore region of Kv11.1 leading to a viable but severe long-QT syndrome
2023-12-21

Congratulations to Antoine, Olga and the team of
Philippe Chevalier (Rhythmology department, HCL Lyon) for our first collaborative study about
Kv11.1/hERG potassium channels.
Highlights
- Homozygous missense variants in the pore of Kv11.1 often lead to intrauterine death.
- We describe a novel hERG p.Gly603Ser homozygous loss-of-function variant.
- It causes a severe but viable long-QT syndrome with a delayed clinical expression.
- Family segregation and functional analysis classify hERG p.Gly603Ser as probably pathogenic.
DYSCO - Dystrophin-associated protein complex and subcellular compartmentalization
2023-07-13

Our project with the team of
Vincent Mirouse (iGred, Clermont-Ferrand) and
Helge Amthor (UVSQ) has been selected by ANR ! The Dystrophin Associated Protein Complex (DAPC) is a key actor of the cell – extracellular matrix (ECM) interface, as revealed by its implication in human genetic disorders. However, its molecular and cellular functions are still poorly understood because tractable model systems allowing state-of-the-art in vivo cell biology and genetic approaches are lacking. Our three teams have developed the first transgenic dystrophin reporters that have revealed remarkably compartmentalized membrane distributions of the DAPC in epithelia and muscle cells in
C. elegans, Drosophila, and mouse. The project aims to elucidate the organization and dynamics of the DAPC and to characterize its new functions in relation to specific cell cortical compartments.
Paper Alert - Distinct dystrophin and Wnt/Ror-dependent pathways establish planar-polarized membrane compartments in C. elegans muscles
2023-03-28

Our study revealing the remarkably complex organisation of the worm's sarcolemma is now on
BioRxiv! Four and half years in the making, congratulations to Alice, Nora, Marie, Amandine, Noémie and Olga for this
magnum opus revealing this entirely unsuspected new case of
planar cell polarity in
C. elegans muscles.
SUMMARY
The plasma membrane of excitable cells is highly structured and molecular scaffolds recruit proteins to specific membrane compartments. Here, we show that potassium channels and proteins belonging to the dystrophin-associated protein complex define multiple types of planar-polarized membrane compartments at the surface of C. elegans muscle cells. Surprisingly, conserved planar cell polarity proteins are not required for this process. However, we implicate a Wnt signaling module involving the Wnt ligand EGL-20, the Wnt receptor CAM-1, and the intracellular effector DSH-1/disheveled in the formation of this cell polarity pattern. Moreover, using time-resolved and tissue-specific protein degradation, we demonstrate that muscle cell polarity is a dynamic state, requiring continued presence of DSH-1 throughout post-embryonic life. Our results reveal the intricate, highly reproducible, and entirely unsuspected complexity of the worm's sarcolemma. This novel case of planar cell polarity in a tractable genetic model organism may provide valuable insight into the molecular and cellular mechanisms that regulate cellular organization, allowing specific functions to be compartmentalized within distinct plasma membrane domains.
Fleggsibility - Dissecting the microevolutionary flexibility of a neural circuit
2022-08-13
Our project with the team of
Christian Braendle (iBV Nice) and
ViewPoint: Behavior Analysis Technologies has been selected by ANR ! On the heals of our joint publication (Vigne et al.,
Science Advances 2021) we will be trying to understand how specific neural circuits evolve to generate natural behavioural variation within species. In particular, the precise genetic changes that modulate cellular and developmental architectures of reproductive systems remain unclear. Here we will focus on the simple egg-laying circuit of the nematode
Caenorhabditis elegans as a powerful model system to study natural microevolutionary (intraspecific) variability.